 Atlantic Yellow-legged Gull (atlantis)
Atlantic Yellow-legged Gull (atlantis)
(last update: October 12, 2011)
Atlantic YLG 4cy April
Below you will find a summary of the Liebers 2001 article published in Molecular Ecology (2001) 10.
"we" in the text below refers to the original authors. If any errors occur in this text, please let me know and mail to marsmuusseatgmaildotcom.
D. LIEBERS, A. J. HELBIG and P. DE KNIJFF
"Genetic differentiation and phylogeography of gulls in the
Larus cachinnans-fuscus group (Aves: Charadriiformes)".
IN: Molecular Ecology (2001) 10, p 2447-2462
Abstract
We studied mitochondrial genetic differentiation among nine taxa of large gulls of the Larus cachinnans-fuscus group, which form part of the circumpolar Herring Gull complex. Our primary interest was to see if there were unrecognized gene flow barriers, to what extent mitochondrial genetic population structure conformed to current taxonomic boundaries, and what it might reveal about possible differences in population history. Sequences (430 nucleotides) of the hypervariable control region I (HVR-I) were obtained from 580 individuals and proved highly informative within this recently diverged group of birds. Contrary to current classification, a basal split was revealed between an Atlantic-Mediterranean clade (atlantis, michahellis, armenicus) and a NW Palearctic-Central Asian clade (cachinnans, barabensis, mongolicus, fuscus·group). There was almost no mitochondrial gene flow between these two groups, although they are in geographical contact in two areas (eastern North Atlantic, Black Sea). Within each of the two major groups, there was strong phylogeographic structure with gene flow barriers between some neighbouring taxa (e.g. cachinnans vs. barabensis), but also a case of poor genetic differentiation between phenotypically distinct forms (barabensis vs. heuglini). At the subspecies level, current taxonomy corresponded well to molecular genetic structure: over 80% of the molecular genetic variance was partitioned among six (groups of) taxa. This is in sharp contrast to previous studies using allozymes and amplified fragment length polymorphism (AFLP) markers, which seemed to indicate extensive nuclear gene flow. Within-taxon haplotype phylogenies and mismatch distributions revealed contrasting demographic histories: cachinnans (Ponto-Caspian region) and atlantis (NE Atlantic) represent ancient lineages with large long-term population sizes, inland forms stem from very recent colonization events (barabensis, mongolicus) or passed through a population bottleneck (armenicus).
Introduction
The authors use an extensive set of mitochondrial HVR-1 sequences to investigate the genetic differentiation and population structure of nine gull taxa, which include all forms of Yellow-legged Gull (cachinnans group) and three forms of Lesser Black-backed Gull (fuscus group) with which the former come into geographical contact. This is the case: (i) in the eastern North Atlantic, where northern, dark-mantled graellsii (breeding on Iceland, Faeroes, British Isles, NW France) meet southern paler-mantled atlantis (Azores, Madeira, Canary Islands) and michahellis along the French Atlantic coast (Yésou 1991); and (ii) along the Ob river depression in western Siberia, where the range of southern barabensis (cachinnans group) approaches that of inland-breeding, northern heuglini (fuscus group).
The primary interest was: (i) to see if there might be unrecognized gene flow barriers; (ii) to what extent mitochondrial genetic population structure conformed to current taxonomic boundaries; and (iii) what it might reveal about possible differences in population history. The authors expect this group of gulls to serve as a suitable model for the many other complexes of closely related forms of birds, which were traditionally classified as polytypic species, but often consist of distinct taxa separated by quite pronounced gene flow.
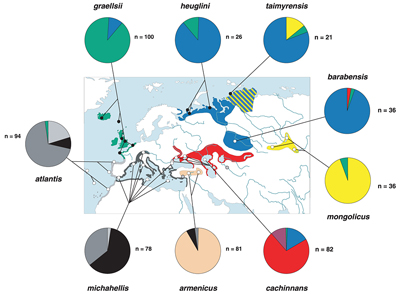 Figure 1.
Figure 1.
Materials and Methods
Sampling
Samples of blood or flight muscle were collected from 580 individuals at 34 localities (Fig. 1), comprising all described taxa in the Larus cachinnans group (atlantis, michahellis, armenicus, cachinnans, barabensis, mongolicus) and three taxa of the L. fuscus group (graellsii, heuglini, taimyrensis). Material was collected exclusively from breeding colonies (see Table 1), either from incubating adults caught with walk-in traps on the nest or from chicks (one per nest). Phenotypic characters of adults (colour of mantle, legs, bill, wing-tip pattern, standard measurements) were recorded at all colonies. Representative voucher specimens and aliquots of all samples investigated in this study have been deposited in the Zoological Museum Greifswald. In phylogenetic analyses, L. canus (from the Netherlands) was used as the outgroup taxon.
Population genetic analyses
For the purpose of this analysis, we pooled individuals of the same taxon from geographically close locations, yielding a total of 22 ‘populations’ (Table 1), each with a minimum sample size of 10. At two sites, Lake Beysehir, SW Turkey, and Tuzkan, Uzbekistan, two distinct phenotypes were breeding in mixed colonies, michahellis and armenicus at the former, cachinnans and barabensis at the latter. Here it was not possible to assign chicks to one or the other taxon (no adults were caught), therefore sequences were excluded from the population genetic analyses. To characterize and compare mitochondrial genetic diversity between taxa, numbers of haplotypes (HT) and polymorphic sites (S), nucleotide diversity (Tc) with variance V(Tc) and mean number of pairwise differences (d) were derived for all taxa using the program ARLEQUIN version 1.1 (Schneider et nl. 1998).
Hierarchical analyses of molecular variance (AMOVA; Excoffier et al. 1992) were performed to study the partitioning of genetic variance within and among populations and taxa. To find the smallest possible number of taxon groups explaining a maximum of between-group genetic variance (i.e. to capture as much of the geographical structure as possible), we assigned the 32 phenotypically pure colonies to two, three, six or nine groups (see ’Results') and estimated the partitioning of genetic variance for each hypothesis using ARLEQUIN.
We computed frequency distributions of pairwise sequence differences (’mismatch distributions') to infer historical demographic patterns within each taxon (Slatkin & Hudson 1991,: Rogers & Harpending 1992) and to calculate per cent sequence divergence between
taxa.
Results
Characterization of sequence variation
The number of haplotypes found per taxon ranged from three (mongolicus) to 25 (cachinnans; Table 2 in PDF). The frequency of each haplotype per population is given in Appendix I (also in PDF). The index of sample saturation (Table 2 in PDF) indicates that most taxa were sampled adequately (values > 1), except heuglini and taimyrensis, for which sample sizes were smallest, and cachinnans, which is by far the most genetically diverse taxon investigated (see below).
Geographical structure of genetic variation
The haplotype composition of the nine phenotypically distinct taxa showed obvious geographical structure (Fig. 1) with pairwise CDST values ranging from 0.032 to 0.928 (Table 3 in PDF). There was significant genetic differentiation between all pairs of taxa except between barabensis and heuglini. In a hierarchical AMOVA, we investigated how the overall genetic variation was distributed among groups of populations (Table 4 in PDF).
Model A: the traditional division into two species, the southern L. cachinnans (pale grey mantle) and the northern L. fuscus (dark grey mantle; cf. Mayr 1963), accounted for only 26.8% of the overall variance.
Further subdividing L. cachinnans, under Model B, into an Atlantic-Mediterranean group (atlantis, michahellis, armenicus) and a West-central Asian group (cachinnans, barbensis, mongolicus) increased the among-groups variance component to 61.5%.
Boundaries between all nine taxa (Model D) explained 81.3% of the variance.
An equally large proportion (82.1%) can be accounted for by only six groups (Model C), if poorly differentiated taxa (CDST values below 0.2; Table 3 in PDF) are merged (atlantis-michahellis and barabensis-heuglini-taimyrensis, respectively). In conclusion, the AMOVA showed that the overall genetic variation has a strong geographical structure and that this structure is well reflected in the current taxonomic subdivisions, which are based on phenotypic characters.
Levels of gene flow in contact zones
An aspect of particular interest was the question to what extent, if any, gene flow between geographically neighbouring taxa might be restricted. This is expressed quantitatively in the QJST values of the AMOVA (Table 3 in PDF) and is illustrated by the relative frequencies of haplotypes in each taxon (Fig. 1). At one extreme, a strong restriction of mitochondrial gene flow was detected: (i) in the eastern North Atlantic between graellsii and atlantis/michahellis; and (ii) on the western coast of the Black Sea between michahellis and cachinnans. In the latter case, no evidence of introgression was found in fairly large samples (n > 80) from the entire range of each taxon. Note that these forms have, until now, been regarded as conspecific. In case (i), two graellsii haplotypes were detected in atlantis breeding colonies (off Portugal and Morocco) indicating that introgression does occur at a very low level. A third contact area with restricted gene flow but higher levels of (unidirectional) introgression is in southern Turkey between michahellis (Mediterranean Sea) and armenicus (Anatolian plateau). The westernmost colony of phenotypically pure armenicus at Tuz Golu contained 14% rnichahellis haplotypes (Liebers & Helbig 1999).
Gene flow between cachinnans and barabensis appeared to be asymmetrical. Although they were significantly differentiated, a considerable proportion (15.8%) of haplotypes typical of barabensis (blue colour in Fig. 1) was found in the cachinnans population, while only 2.8% cachinnans haplotypes were found in barabensis. Unexpectedly, barabensis and heuglini turned out not to be differentiated at the mitochondrial genetic level (Table 3 in PDF), although they clearly differ in mantle colour and, according to current knowledge, their ranges do not come into contact. The degree of gene flow restriction was not obviously related to topographical barriers: no such barriers separate michahellis from cachinnans on the Black Sea coast (no gene flow) or graellsii from atlantis/michahellis on the Atlantic coast and islands (very little introgression). On the other hand, michahellis and armenicus (more extensive introgression) are separated by the Taurus mountains (up to 3000 m high), inhospitable terrain for large gulls which, however, has not prevented michahellis from wandering into armenicus territory at least occasionally.
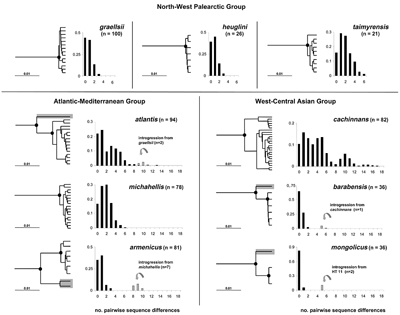 Figure 2.
Figure 2.
Population history and haplotype phylogeny within taxa
To investigate possible differences in the demographic history, we analysed the following parameters in each of the nine taxa:
1 Nucleotide diversity and expansion coefficient (Table 2 in PDF).
2 Branching pattern (Fig. 2) and deepest divergence (maximum K 2-p distance, Table 2 in PDF) of the haplotype tree.
3 Mismatch distribution (Fig. 2); a unimodal distribution indicates recent exponential population growth, while a multimodal distribution characterizes a large population that has been of relatively constant size over time (Rogers & Harpending 1992).
ln atlantis, armenicus, barabensis and mongolicus a low proportion of phylogenetically distantly related haplotypes was found which, in all probability, reflect recent introgression (highlighted by grey shading in Fig. 2). To assess the impact of such introgression on the population genetic parameters, we calculated them separately with and without introgressed haplotypes in the four taxa concerned (Table 2 in PDF).
With regard to genetic diversity and haplotype divergence patterns, two extremes can be distinguished: (i) taxa with a shallow branching pattern of their haplotype tree corresponding to a strongly left-skewed, unimodal mismatch distribution and limited genetic diversity (graellsii, heuglini, barabernsis, armenicus, mongolicus);
and (ii) taxa with a multimodal mismatch distribution, a correspondingly deep branching pattern of the haplotype phylogeny and high genetic diversity (atlantis, cachinnans). Two taxa had a fairly broad, but unimodal, mismatch distribution and intermediate depth of branching pattern (taimyrensis, michahellis). Pairwise comparisons showed that cachinnans had significantly higher nucleotide diversity than all other taxa except atlantis and taimyrensis.
Historical changes in population size can be assessed by the ’expansion coefficient’ S / d, i.e. the ratio of variable sequence positions (S) relative to the mean number of pairwise nucleotide differences between haplotypes (d) within a taxon. Large values indicate recent population expansion, whereas the ratio will be small in populations that have been constant in size (von Haeseler et al. 1996). S/ d ratios ranged from 5.3 to 17.8 with highest values in graellsii, mongolicus and barabensis (Table 2 in PDF). The latter three also exhibit a shallow haplotype phylogeny (Fig. 2), together strongly indicating recent population expansion. Among the other taxa, the S / d ratio varied little (5.3-7.6), suggesting that their populations have been relatively constant in size over long periods of time. The Armenian Gull (armenicus), although representing a phylogenetically relatively old lineage (see below), had an intermediate S / d value of 9.5, if introgressed haplotypes were disregarded. This indicates that the population went through a bottleneck relatively recently.
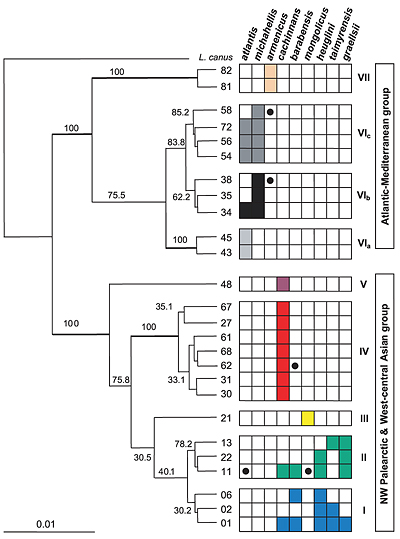 Figure 3.
Figure 3.
mtDNA haplotype phylogeny
Of the total of 90 haplotypes, a large fraction (71%) was
confined to single breeding colonies (or colonies in close
proximity). We restrict our analysis of the mtDNA
phylogeny to those 26 haplotypes that were shared
between at least two populations (see Appendix I in PDF), because
these contain most information about relationships of
mtDNA lineages and taxa. The molecular clock hypothesis
was not rejected in a likelihood ratio test (puzzle), i.e. rates of molecular evolution did not differ significantly between
lineages.
The haplotype phylogeny (Fig. 3) reveals a strongly
supported basal split into two clades, one occurring
exclusively in the Atlantic / Mediterranean group of gulls
(atlantis, michahellis, armenicus), the other being largely
restricted to a NW Palearctic / Central Asian group. None
of the nine taxa were represented by a monophyletic clade
of haplotypes relative to all other forms. In some cases this
was due to obvious recent introgression, such as the two
michahellis haplotypes (nos 38, 58) found in armenicus, one
cachinnans haplotype (62) found in barabensis and one clade
II haplotype (11) found in atlantis and mongolicus (marked
by dots in grid of Fig. 3). In other cases the lack of reciprocal monophyly may be due to incomplete lineage sorting
between very recently diverged forms (heuglini, taimyrensis,
barabensis) and/or ongoing gene flow (e.g. atlantis,
michahellis).
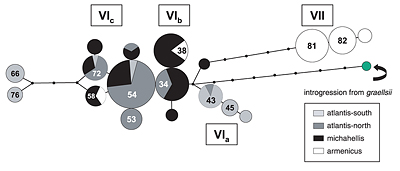 Figure 4.
Figure 4.
The Atlantic / Mediterranean clade exhibits a somewhat clearer phylogenetic structure (vs the NW Palearctic / Central Asian group): haplotypes found in atlantis and michahellis, some of which are shared between them, together make up a well-supported monophyletic group (clades VIa-c in Fig. 3). Armenicus was found to be their sister taxon and, notwithstanding its shallow current branching pattern (see Fig. 2), represents a fairly ancient haplotype lineage (clade VII) with some, apparently recent, introgression from michahellis.
Population genetic structure of 'Yellow-legged Gulls'
We have shown that the six gull taxa of the southern
Palearctic fall into two major groups, between which no
mitochondrial gene flow was detected:
(i) Atlantic /
Mediterranean group: atlantis, michahellis, armenicus; and
(ii) West-central Asian group: cachinnans, barabensis,
mongolicus.
We will look at the population genetic structure
within each of these groups in some more detail.
Atlantic / Mediterranean group. A median-joining network
(Fig. 4a) shows that mitochondrial haplotypes occurring in
the michahellis population are a subset, or are recently
derived from, those found in atlantis. Michahellis also has
a more shallow haplotype phylogeny and lower nucleotide
diversity than atlantis. Within the michahellis range thereis no significant geographical substructure, whereas there is
significant differentiation between most of the five atlantis colonies analysed (Table 5a in PDF). Among gulls of the Atlantic
islands and mainland coasts we found an unexpected
north-south pattern of differentiation: the Azores population
was similar to that of the Iberian Atlantic coast (1350 km
away), but was significantly differentiated from the
Madeiran (900 km distant) and mainland Moroccan breeding
birds (Table 5a in PDF). Since most michahellis haplotypes are
either identical or closely related to those of "northern"
(Azores, mainland Iberia) rather than "southern" (Madeira,
Morocco) atlantis populations, it seems that the latter
contributed few colonizers to the Mediterranean.
Discussion
Phylogenetic relationships
Earlier attempts based on phenotypic characters
(Stegmann 1934; Johansen 1960; Chu 1998) allozymes (Johnson 1985; Snell 1991), AFLP markers (de Knijff et al.
2001) and some mtDNA sequences (Heidrich et al. 1996;
Crochet et al. 2000) achieved only poor resolution of
the phylogenetic relationships, suggesting that these large
gulls derive from a relatively rapid radiation. HVR-I
sequences, however, yielded surprisingly clear phylogenetic
information, which greatly improves our understanding of relationships in this group of birds. Interestingly, the
deepest split in the mitochondrial phylogeny did not
separate northern dark-mantled (heuglini, taimyrensis, graellsii) from southern light-mantled taxa (all others),
which current taxonomy regards as two different species
(Lesser Black-backed Gull vs. Yellow-legged Gull). Instead,
the basal split was within the southern taxa, separating the Atlantic / Mediterranean atlantis/michahellis from the Aralo /
Caspian cachinnans/barabensis, all of which were so far
regarded as the same species (L. cachinnans).
The Armenian Gull (armenicus), which had been split
off as separate species by Haffer (1982), but whose phylogenetic
affinities were unclear, was firmly placed in the
Atlantic / Mediterranean clade. It branched off the atlantis/michahellis lineage rather basally, indicating that armenicus is a relict of an early colonization event from the Atlantic
via the Mediterranean Basin, rather than from the Aralo /
Caspian region as had previously been thought (Buturlin
1934; Filchagov 1993). Also phenotypically very similar to,
but geographically separated from armenicus, is the west
Siberian barabensis, whose affinities have always been
controversial. We found it to be most closely related to heuglini,
which is supported by evidence from a recent field
and museum study (Panov & Monzikov 2000). Gulls
inhabiting the Azores and Madeira (atlantis) were originally
thought to be part of the fuscus-group (Dwight 1922),
i.e. most closely related to graellsii, because of their relatively
dark mantle and extensive head streaking in adult
nonbreeding plumage (features they share with graellsii).
However, atlantis and graellsii belong to different major
clades in the mitochondrial haplotype phylogeny (Fig. 3)
and are clearly not each other's closest relatives.
Population history
Pleistocene glacial cycles and associated ecological
changes undoubtedly affected the population dynamics of
gulls. Two extremes with respect to population history
were evident among the gull taxa in our study. Genetic
characteristics of cachinnans and atlantis indicate that these
lineages had large populations over long periods. Both
taxa reside today in areas of relative climatic stability: in
the eastern North Atlantic Ocean (atlantis), glacial cycles at
most may have led to north-south range shifts, but not to
severe population bottlenecks or range restrictions. In the
Aralo / Caspian / Pontic region (cachinnans), large inland
seas have persisted throughout the Holocene, probably always providing habitat for large gull populations
(Dawson 1992; Rutter 1995). Two lines of evidence suggest
that cachinnans is a direct descendant of the ancestral
population, from which barabensis, heuglini, taimyrensis,
graellsii and mongolicus are derived:
first, cachinnans has
the deepest divergence in the haplotype tree, the highest
nucleotide diversity and the smallest expansion coefficient
of all taxa investigated, consistent with a large long-term
population size (Slatkin & Hudson 1991; von Haeseler
et al. 1996).
Second, in the haplotype phylogeny (Fig. 3),
cachinnans is paraphyletic relative to the other five taxa.
This pattern is reminiscent of the global phylogeography of
human mtDNA, where African populations contain the
most divergent haplotypes and are paraphyletic relative to
populations in the rest of the world (Ingman et al. 2000).
At the opposite extreme are several taxa that harbour
little mitochondrial genetic diversity and show evidence
of recent population expansion. In barabensis and mongolicus this seems to be due to very recent colonization of inland
areas from different source populations: barabensis was
found not to be differentiated from heuglini, suggesting
that it colonized its present range from the north via the
Ob-Irtys river system. Phenotypic divergence between
heuglini and barabensis (mostly in mantle colour) must have
been fairly rapid, not allowing for measurable mitochondrial
genetic differentiation to accumulate. In mongolicus,
the extreme paucity of genetic variation and a high expansion
coefficient also suggest very recent immigration and
population increase. Haplotypes dominating (at 94%)
throughout its range are related to or identical with those
of eastern Siberian and Pacific gull taxa (vegae, schistisagus;
data not shown), while one haplotype (6% frequency) is
common in heuglini–taimyrensis. This suggests that Central
Asia was colonized primarily from an eastern Siberian
and/or NW Pacific source, probably by relatively few
individuals.
The Armenian Gull (armenicus) is a good example of a
taxon that must have passed through a population bottleneck.
Its current population genetic make-up differs little
from that of recent colonizers such as graellsii or barabensis,
but in contrast to the latter, armenicus derives from a
phylogenetically relatively old lineage (Fig. 3). Had its
population been large over long periods, a much more
diverse and deeply branching haplotype assortment would
be expected. The discrepancy is particularly striking compared
to its sister lineage, the atlantis/michahellis clade (Fig. 3).
Within the atlantis–michahellis group, the haplotype
phylogeny and the decline in nucleotide diversity from the
Atlantic (atlantis) toward the Mediterranean (michahellis)
suggest that Atlantic populations were ancestral to those
living today in the Mediterranean Basin (with peripheral
extensions into Black Sea and inland SW Europe). Consistent
with this hypotheses, michahellis has a shallower haplotype
branching pattern than atlantis and a unimodal, rather than bimodal, mismatch distribution. Gene flow between
these poorly differentiated taxa was estimated to be moderate,
which is to be expected given the continuous oceanic
connection via the Straits of Gibraltar. Interestingly, Mediterranean
michahellis have recently recolonized the French
Atlantic coast via southern France (Yésou 1991), further
increasing the likelihood of genetic exchange with atlantis.
This will oppose further differentiation and lineage sorting
between atlantis and michahellis. On the other hand, the
differentiation of southern (Madeira, Morocco) from northern
(Azores, Iberia) atlantis and from Mediterranean michahellis populations is noteworthy. Among these three, southern
atlantis contain the greatest diversity and largest divergence
of haplotypes (Fig. 4a), suggesting that southern
populations were more stable throughout periods of
glacial oscillations. It is not obvious what may today restrict gene flow between the oceanic island groups (Madeira,
Azores), or why the southern atlantis population evidently
contributed few colonizers to the Mediterranean.
Age of gull lineages
Dating the split between the two major clades of gulls
identified in this study (Fig. 3) is difficult, because rates
of HVR-I evolution have not been calibrated accurately
in gulls or other Charadriiform birds. More reliable
calibrations are available for the cytochrome b gene
(average 2% divergence per 1 Myr; Avise 2000). Average
cyt b divergence between michahellis and graellsii is 0.007
(data not shown). The mean divergence of HVR-I
sequences between the two major clades is 0.060% (K 2-p
distance with gamma correction). Thus, HVR-I seems to
evolve roughly 8.6 times faster than cyt b, yielding a
divergence rate of 17% per Myr. This calibration dates the
basal split between the major mitochondrial lineages at roughly 350 000 years ago. The separation into two
ancestral populations from which the two major clades of
gull taxa derived must be younger, because lineage
divergence usually predates population-level divergence
(Avise 2000). To date the latter, we need to correct the
divergence estimate for sequence diversity in the ancestral
population as suggested by Edwards (1997). Using as a
correction the mean of current intrapopulation divergence
in atlantis–michahellis (0.0127) and in the cachinnans group
(0.0202), we arrive at a divergence between clades of
d = 0.0436, which yields an age of 256 000 - 295 000 years
for the population-level separation. This separation most
likely corresponded to the reciprocal isolation of gulls in an
Atlantic and a Pontic-Caspian refugium, because most of
the current within-population genetic diversity is localized
in these two regions (atlantis and cachinnans populations).
The cause of the isolation of gulls in two separate refugia
may have been a glacial maximum around 250 000 -
270 000 years bp (Schrag 2000).
.jpg) Atlantic YLG (atlantis) sub-adult, sub-adult, April 06 2008, Funchal, Madeira Islands. Picture: Thijs Valkenburg.
Atlantic YLG (atlantis) sub-adult, sub-adult, April 06 2008, Funchal, Madeira Islands. Picture: Thijs Valkenburg..jpg) Atlantic YLG (atlantis) sub-adult, sub-adult, April 06 2008, Funchal, Madeira Islands. Picture: Thijs Valkenburg. Adult-like plumage, but dark markings on greater primary coverts.
Atlantic YLG (atlantis) sub-adult, sub-adult, April 06 2008, Funchal, Madeira Islands. Picture: Thijs Valkenburg. Adult-like plumage, but dark markings on greater primary coverts..jpg) Atlantic YLG (atlantis) sub-adult, sub-adult, April 06 2008, Funchal, Madeira Islands. Picture: Thijs Valkenburg.
Atlantic YLG (atlantis) sub-adult, sub-adult, April 06 2008, Funchal, Madeira Islands. Picture: Thijs Valkenburg.